5
More Annotations






5



2
Favourite Annotations





5


3
Text
THE AMBER MOLECULAR DYNAMICS PACKAGEDOWNLOAD AMBERINSTALLATIONNEWSAMBER CITATIONSGPU SUPPORTINTEL SUPPORT Welcome to Amber! Amber is a suite of biomolecular simulation programs. It began in the late 1970's, and is maintained by an active development community; see our history page and our contributors page for more information.. The term "Amber" refers to two things.
AMBERTOOLS21
AmberTools21 (released on April 29, 2021) consists of the following major codes: NAB/sff: a program build molecules, run MD or apply distance geometry restraints using generalized Born, Poisson-Boltzmann or 3D-RISM implicit solvent models. antechamber and MCPB: programs to create force fields for general organic molecules and metal centers. AMBER ADVANCED TUTORIALS (Note: These tutorials are meant to provide illustrative examples of how to use the AMBER software suite to carry out simulations that can be run on a simple workstation in a reasonable period of time. RE: COMPILING AMBER WITH GCC 9.2 AND INTEL MPI From: David A Case Date: Mon, 28 Oct 2019 14:48:46 -0400 On Mon, Oct 28, 2019, Vlad Cojocaru wrote: > >I am trying to compile Amber 18 with AmberTools 19 on a supercomputer. >I tried using the default environment that provides intel mpi 2019 and IMPORTERROR: COULD NOT IMPORT AMBER PYTHON MODULES From: Renato Araujo Date: Thu, 9 Apr 2020 14:24:18 -0300 Dear Amber users I am trying to reproduce the tutorial on the pagehttps://ambermd
RE: HOW TO CALCULATE COORDINATION NUMBER FROM RDF This message: ; Next message: Matias Machado: "Re: RESOLVED: peek_ewald_inpcrd: Box info not found in inpcrd"; Previous message: Matias Machado: "Re: unwrap problem in the liquid water dynamics trajectory analysis"; In reply to: Bharat Manna: " How to calculate Coordination number from RDF Profile usingCPPTRAJ ?"
RE: RESTRAINTS NOT WORKING, WHAT AM I DOING WRONG From: Dr. Anselm Horn Date: Tue, 15 Nov 2016 09:45:07 +0100 Dear The Cromicus Productions, the flag ntr=1 is necessary to tell sander that there are restraints, which should be taken into account. RE: BAD TOPOLOGY FILE. SUM OF ATOMS_PER_MOLECULE This message: ; Next message: David A Case: "Re: Bad topology file.Sum of ATOMS_PER_MOLECULE does not equal NATOM" Previous message: Saeed Nasiri: " Bad topology file.Sum of ATOMS_PER_MOLECULE does not equal NATOM" In reply to: Saeed Nasiri: " Bad topology file.Sum of ATOMS_PER_MOLECULE does not equalNATOM"
RE: FATAL: ATOM DOES NOT HAVE A TYPE. FROM From: Jonathan Gough Date: Fri, 7 Dec 2012 16:39:28 -0500 I figured it out I didn't realize there was a connection between the variable name (in tleap) and the name of the molecule in mol2 and the pdb RE: FILE CONTAINS 2 ATOMS NOT IN RESIDUE TEMPLATES From: Jason Swails Date: Tue, 23 Sep 2014 09:46:33 -0400 On Tue, 2014-09-23 at 18:52 +0530, Vishal Nemaysh wrote: > *Dear all, I amgetting a
THE AMBER MOLECULAR DYNAMICS PACKAGEDOWNLOAD AMBERINSTALLATIONNEWSAMBER CITATIONSGPU SUPPORTINTEL SUPPORT Welcome to Amber! Amber is a suite of biomolecular simulation programs. It began in the late 1970's, and is maintained by an active development community; see our history page and our contributors page for more information.. The term "Amber" refers to two things.AMBERTOOLS21
AmberTools21 (released on April 29, 2021) consists of the following major codes: NAB/sff: a program build molecules, run MD or apply distance geometry restraints using generalized Born, Poisson-Boltzmann or 3D-RISM implicit solvent models. antechamber and MCPB: programs to create force fields for general organic molecules and metal centers. AMBER ADVANCED TUTORIALS (Note: These tutorials are meant to provide illustrative examples of how to use the AMBER software suite to carry out simulations that can be run on a simple workstation in a reasonable period of time. RE: COMPILING AMBER WITH GCC 9.2 AND INTEL MPI From: David A Case Date: Mon, 28 Oct 2019 14:48:46 -0400 On Mon, Oct 28, 2019, Vlad Cojocaru wrote: > >I am trying to compile Amber 18 with AmberTools 19 on a supercomputer. >I tried using the default environment that provides intel mpi 2019 and IMPORTERROR: COULD NOT IMPORT AMBER PYTHON MODULES From: Renato Araujo Date: Thu, 9 Apr 2020 14:24:18 -0300 Dear Amber users I am trying to reproduce the tutorial on the pagehttps://ambermd
RE: HOW TO CALCULATE COORDINATION NUMBER FROM RDF This message: ; Next message: Matias Machado: "Re: RESOLVED: peek_ewald_inpcrd: Box info not found in inpcrd"; Previous message: Matias Machado: "Re: unwrap problem in the liquid water dynamics trajectory analysis"; In reply to: Bharat Manna: " How to calculate Coordination number from RDF Profile usingCPPTRAJ ?"
RE: RESTRAINTS NOT WORKING, WHAT AM I DOING WRONG From: Dr. Anselm Horn Date: Tue, 15 Nov 2016 09:45:07 +0100 Dear The Cromicus Productions, the flag ntr=1 is necessary to tell sander that there are restraints, which should be taken into account. RE: BAD TOPOLOGY FILE. SUM OF ATOMS_PER_MOLECULE This message: ; Next message: David A Case: "Re: Bad topology file.Sum of ATOMS_PER_MOLECULE does not equal NATOM" Previous message: Saeed Nasiri: " Bad topology file.Sum of ATOMS_PER_MOLECULE does not equal NATOM" In reply to: Saeed Nasiri: " Bad topology file.Sum of ATOMS_PER_MOLECULE does not equalNATOM"
RE: FATAL: ATOM DOES NOT HAVE A TYPE. FROM From: Jonathan Gough Date: Fri, 7 Dec 2012 16:39:28 -0500 I figured it out I didn't realize there was a connection between the variable name (in tleap) and the name of the molecule in mol2 and the pdb RE: FILE CONTAINS 2 ATOMS NOT IN RESIDUE TEMPLATES From: Jason Swails Date: Tue, 23 Sep 2014 09:46:33 -0400 On Tue, 2014-09-23 at 18:52 +0530, Vishal Nemaysh wrote: > *Dear all, I amgetting a
THE AMBER MOLECULAR DYNAMICS PACKAGE The term "Amber" refers to two things. First, it is a set of molecular mechanical force fields for the simulation of biomolecules (these force fields are in the public domain, and are used in a variety of simulation programs). Second, it is a package of molecular simulation programs which includes source code and demos.AMBER TUTORIALS
Amber Tutorials. Below are tutorials prepared by the Amber developers to help you learn how to use the Amber software suite. There are also two sets of Japanese versions: one from Akira Wadano and Takenori Kanai, and a second prepared by Conflex corporation. **Please note that some of these tutorials were written to run quickly for illustrative purposes. PLACING EXTRA POINTS WITH MDGX Placing Customized Extra Points in an Amber Topology File. The mdgx &parmedit module serves a purpose with overlap in parmed and tleap, and while those programs could, in time, take over this functionality it was expedient to have the feature in mdgx as there is no file format precedent for it. Extra points, or "virtual sites" are a powerful way to improve the monopole distribution of a molecule. 1.5 CALCULATING SALT MOLARITY IN AN EXPLICIT WATER SYSTEM 1.5 Calculating Salt Molarity in an Explicit Water System. By Abigail Held 1 and Maria Nagan 2 1 from Bill Miller III's lab, Truman State University, 2 Stony Brook University. Learning Outcomes; Obtain the volume from LEaP; Calculate the number of buffer ions to add in LEaP based on an overall molarity GENERATING FORCE FIELD PARAMETERS WITH PARAMFIT The first step is to create a topology file containing the parameters to be fit. For our system, the dihedrals of interest are defined to contain only two terms, as found in parm99.dat: N -CT-C -N 1 1.700 180.000 -1. N -CT-C -N 1 2.000 180.000 2. C -N -CT-C 1 0.850 180.000-2.
ANTECHAMBER AND GAFF CITATIONS Wang, J., Wang, W., Kollman P. A.; Case, D. A. "Automatic atom type and bond type perception in molecular mechanical calculations". Journal of Molecular Graphics and RE: RESTRAINTS NOT WORKING, WHAT AM I DOING WRONG From: Dr. Anselm Horn Date: Tue, 15 Nov 2016 09:45:07 +0100 Dear The Cromicus Productions, the flag ntr=1 is necessary to tell sander that there are restraints, which should be taken into account. RE: AT LINE 434 OF FILE MAKE_CRD_HG.F (UNIT = 5 From: David A Case Date: Thu, 26 Nov 2015 15:07:46 -0700 On Thu, Nov 26, 2015, leila karami wrote: > > I am using mmpbsa.pl in extracting RE: PROBLEMS WITH MMPBSA FROM JASON SWAILS ON 2014 an atom is. It is used to construct the boundary between the internal. dielectric of the substance (1 to 2) and the external dielectric of the. solvent (~80 for water). PB and GB construct this dielectric boundary. differently, but both require the size of each atom asinput. This.
RE: AMBPDB COULD NOT READ RESTART ATOMS/TIME. FROM From: David A Case Date: Thu, 10 Mar 2016 07:14:10 -0500 On Wed, Mar 09, 2016, Dan Roe wrote: > > Try using cpptraj to convert the restart . Note that Bin is already using the cpptraj code:AmberTools20
Amber20
Manuals
Tutorials
Force Fields
Contacts
History
USEFUL LINKS:
Download Amber
Installation
News
Amber Citations
GPU Support
Intel Support
Updates
Mailing Lists
For Educators
File Formats
Custom Search
WELCOME TO AMBER!
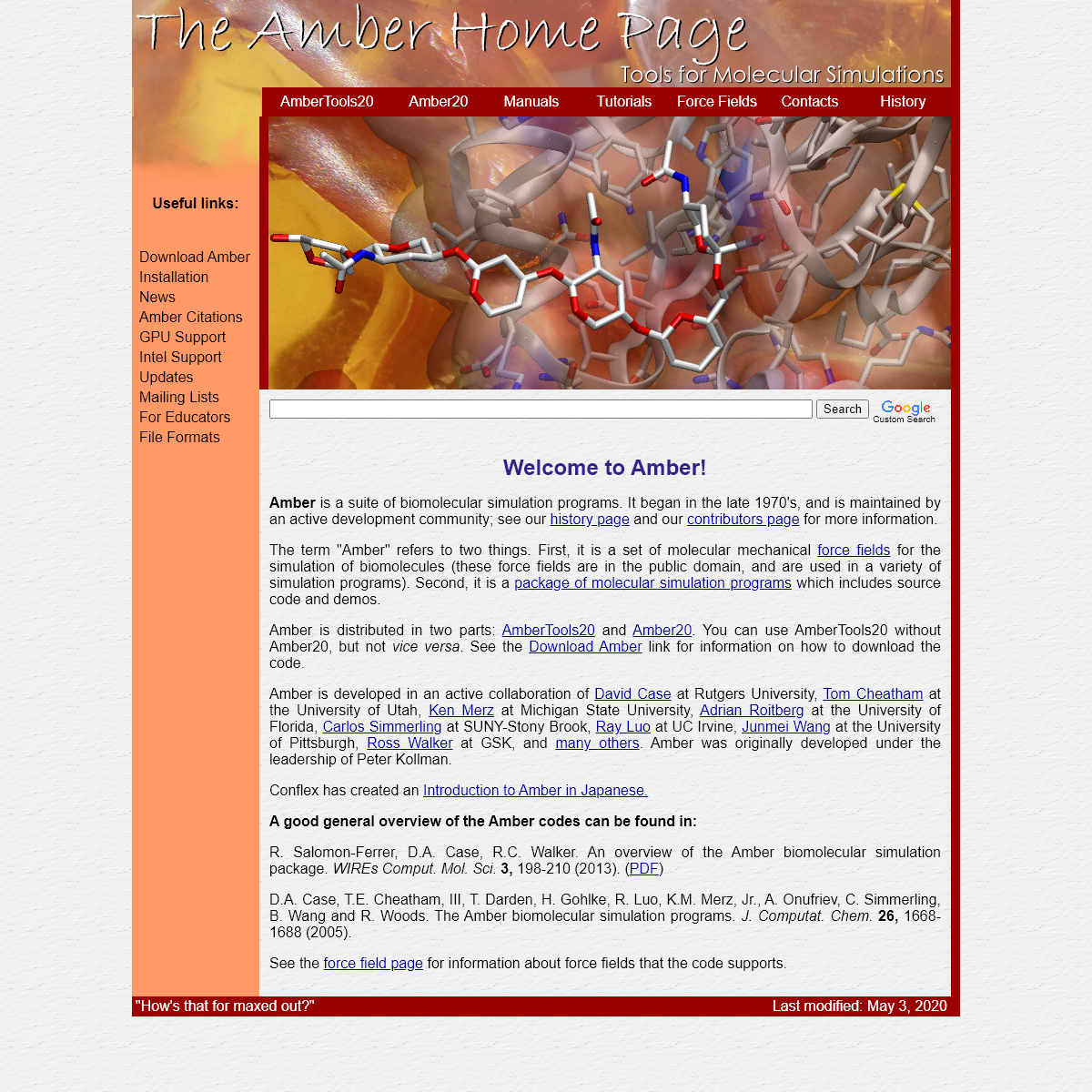
AMBER is a suite of biomolecular simulation programs. It began in the late 1970's, and is maintained by an active development community; see our history page and our contributors page for more information. The term "Amber" refers to two things. First, it is a set of molecular mechanical force fields for the simulation of biomolecules (these force fields are in the public domain, and are used in a variety of simulation programs). Second, it is a package of molecular simulation programs which includes sourcecode and demos.
Amber is distributed in two parts: AmberTools20 and Amber20 . You can use AmberTools20 without Amber20, but not _vice versa_. See the Download Amber link for information on how to download the code. Amber is developed in an active collaboration of David Case at Rutgers University, Tom Cheatham at the University of Utah, Ken Merz at Michigan State University, Adrian Roitberg at the University of Florida, Carlos Simmerlingat SUNY-Stony
Brook, Ray Luo
at UC
Irvine, Junmei Wang
at
the University of Pittsburgh, Ross Walker at GSK, and many others . Amber was originally developed under the leadership of Peter Kollman. Conflex has created an Introduction to Amber in Japanese. A GOOD GENERAL OVERVIEW OF THE AMBER CODES CAN BE FOUND IN: R. Salomon-Ferrer, D.A. Case, R.C. Walker. An overview of the Amber biomolecular simulation package. _WIREs Comput. Mol. Sci._ 3, 198-210(2013). (PDF )
D.A. Case, T.E. Cheatham, III, T. Darden, H. Gohlke, R. Luo, K.M. Merz, Jr., A. Onufriev, C. Simmerling, B. Wang and R. Woods. The Amber biomolecular simulation programs. _J. Computat. Chem._ 26, 1668-1688(2005).
See the force field page for information about force fields that the code supports. "How's that for maxed out?" Last modified: May 3, 2020Details
1